

Hannes Steinkellner, Priv.-Doz. Mag. Dr.
Principal InvestigatorTel.: +43 (0)1 40160-56534
E-Mail: hannes.steinkellner@meduniwien.ac.at
Research focus
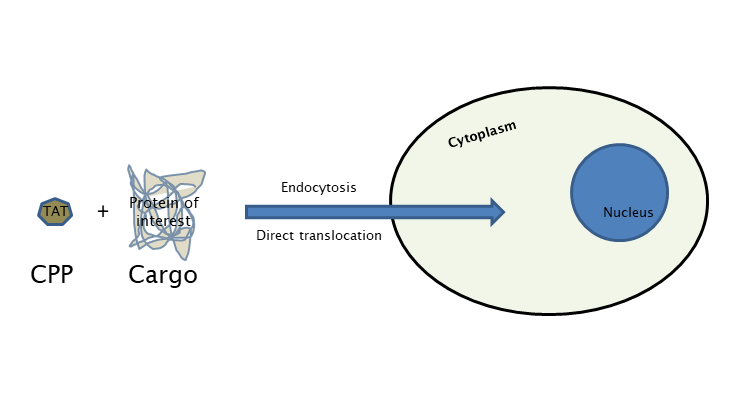
The main research focus of Laccone Lab is the development and investigation of TAT (transactivator of transcription)- fusion proteins for neurodevelopmental and neurodegenerative disorders like RETT syndrome, Spinal Muscular Atrophy and Friedreich’s Ataxia. The technology requires the synthesis of the gene encoding for a fusion protein, linking the TAT transduction domain to the molecule of interest using a bacterial expression vector, followed by the purification of this fusion protein under either soluble or denaturing conditions. The purified fusion protein can be directly added to mammalian cell culture or injected in vivo into mice. Full-length TAT fusion proteins have been used to address a number of biological questions, relating to cell cycle progression, apoptosis, and cellular architecture.
The Laccone Lab is using methods such as Dynamic Light Scattering (DLS), Small Angle X-Ray Scattering (SAXS) and Nuclear Magnetic Resonance (NMR) spectroscopy to investigate protein stability (both conformational and otherwise), aggregation propensity and the presence of folded domains. Stable, non-aggregating protein constructs at optimal buffer conditions yield more favorable results in downstream assays, such as cell culture experiments
The application of a recombinant protein for a possible treatment of RETT syndrome has been patented: “Synthetic MECP2 sequence for protein substitution therapy“, (WO/2007/115578). However, we also attempt to develop and investigate recombinant proteins as a possible strategy for other genetic diseases.
Furthermore, Franco Laccone is interested in recruitment of patients with unknown illnesses for their identification.
Development of various TAT-fusion proteins for rare diseases like RETT syndrome, Spinal muscular atrophy and Friedreich’s ataxia
- Expression, purification and structural characterization of MeCP2 constructs
- Drug delivery across the blood-brain barrier for protein replacement therapeutic approaches against Rett syndrome
- Changes of the blood-brain barrier during RETT syndrome
- The role of MeCP2 in the interaction within the neurovascular unit
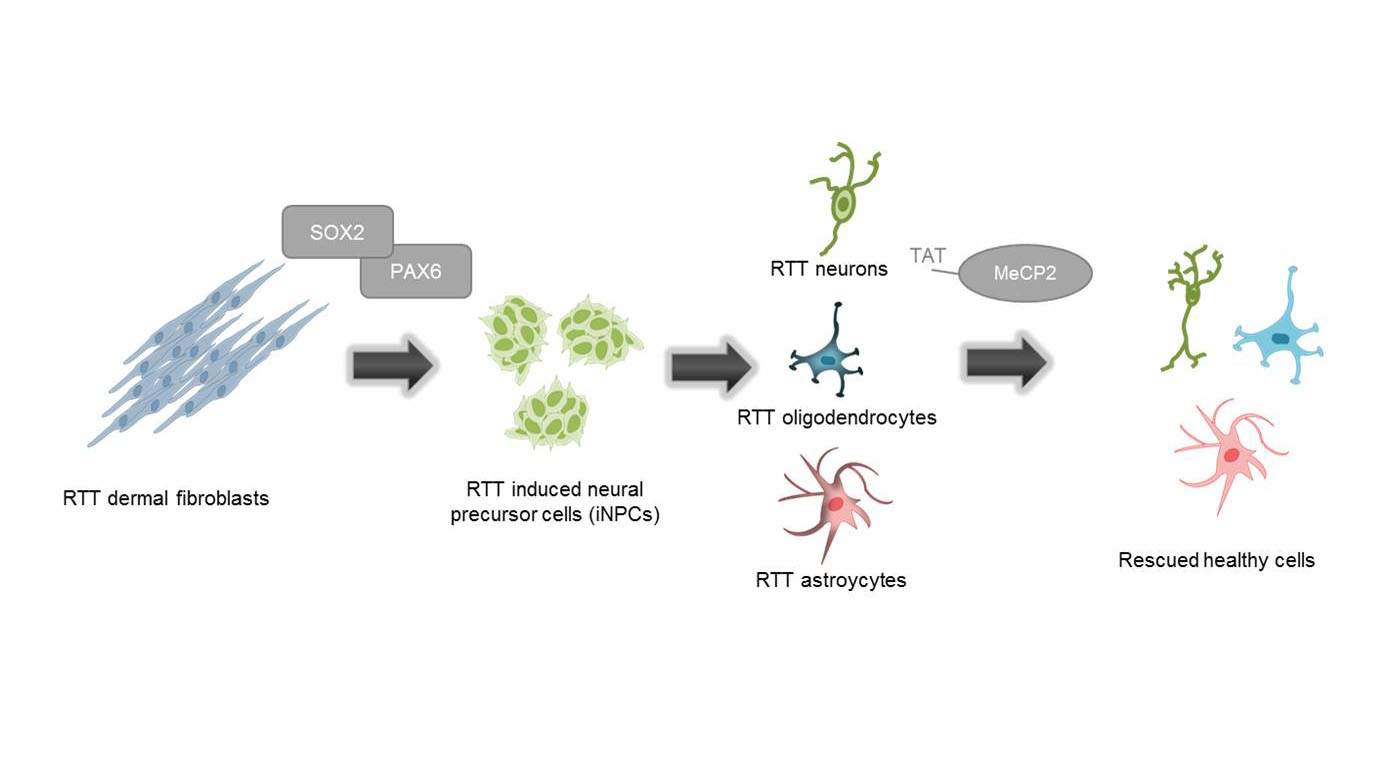
Generation of functionally active neurons using direct conversion from RETT patient fibroblast
Human fibroblasts from a MeCP2-deficient patient (and a healthy wildtype) are transfected with two episomal plasmids which encode for the transcription factors PAX6 and SOX2. Transfected cells are cultured in reprogramming medium for several weeks, resulting in induced neuronal progenitor cells (iNPs), which are in turn differentiated into neurons. This patient-derived cell model for RTT could serve as a promising tool for investigating phenotype rescue through the delivery of a recombinant MeCP2 protein.
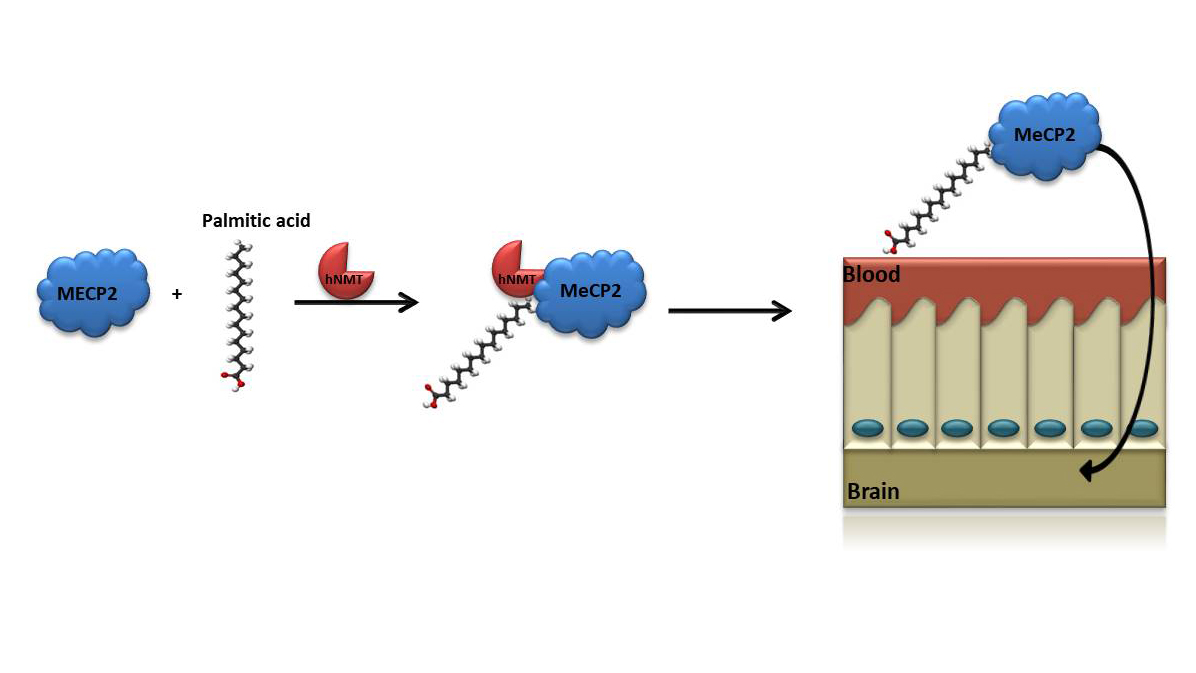
Monoacylation of recombinant proteins for crossing the blood-brain barrier
A major challenge in correcting disorders like RETT syndrome is to induce BBB crossing of exogenously applied compounds.
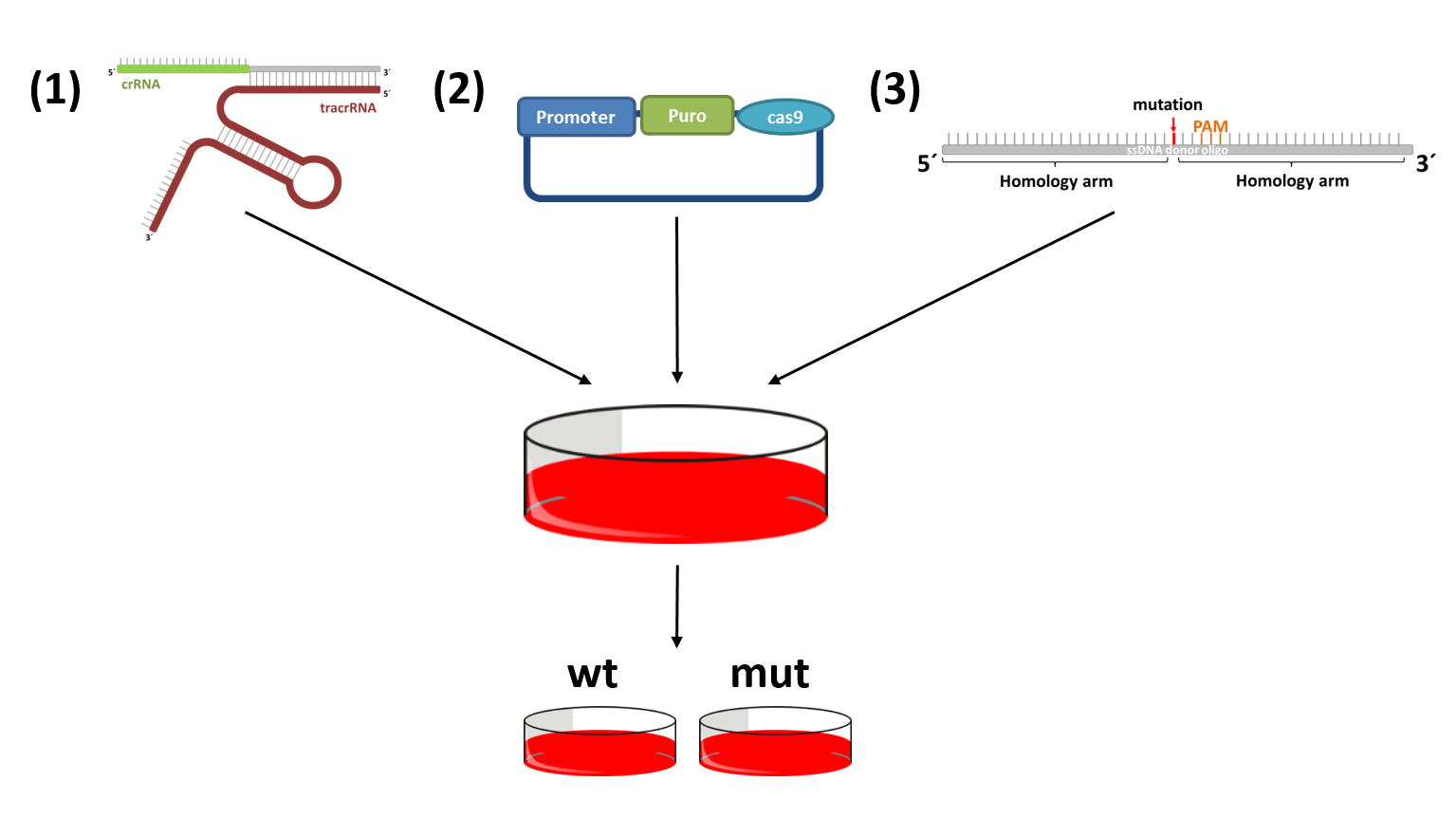
Functional characterization of various candidate genes by CRISPR/Cas9 mediated genome editing
The CRISPR-cas9 system can be used to induce mutations that have previously been observed in patients. This allows the direct comparison of cells with a wildtype and mutant gene of interest isolated form other influences. CRISPR-cas9 makes use of a double strand break repair mechanism of mammalian cells, homology directed repair. There are only three crucial parts: (1) cas9, to induce a double strand break; (2) a site specific sgRNA or crRNA:tracrRNA complex that recruits cas9; and (3) a donor DNA harboring the desired mutation.
- AIRETT – Associazione Italiana Rett (currently)
- Elternhilfe für Kinder mit RETT Syndrome (from 2006 - 2016)
- The Ludwig Boltzmann Institute of Osteology (LBIO)
Thomas Dechat, PhD - Austrian Institute of Technology
Winfried Neuhaus, Priv.-Doz.
Dipl.Ing. Dr., AIT - Center of Brain Research
Sigismund Huck, Prof.
Petra Scholze, Assoc.Prof., MedUni Vienna - Centre for Brain Research - the University of Auckland
Bronwen Connor, Prof.
Current Lab Members

Hannes Steinkellner, Priv.-Doz. Mag. Dr.
Principal InvestigatorTel: +43 (0)1 40160-56534
E-Mail: hannes.steinkellner@meduniwien.ac.at
PubMed, Research gate, Researcher profile


Former Members of Laccone Lab
-
Teresa Seipel (Master Student)
-
Anna Huber (PhD Student)
-
Claudia Sulek (Master Student)
-
Alexander Weiss (Master Student)
-
Sofia Geislberger (Bachelor Student)
-
Melanie Olczykowski (Master Student)
-
Mara Kluge (Diploma Student)
-
Philip Mausberg (Master Student)
-
Fabian Huber (Diploma Student)
-
Julia Etzler (Technician)
-
Katrin Rose (Master Student)
-
Neli Bounzina (Master Student)
-
Alexander Reitner (Master Student)
-
Zsofia Kormanyos (Master Student)
-
Azra Kurtovic (Master Student)
-
Anna Schönegger (Master Student)
-
Laura Gogoll (Diploma student)
-
Pinar Kehrer (Master Student)
-
Sandra Pferschy (Postdoc)
-
Gerwin Heller (Postdoc)
-
Generation and Characterization of a Human Neuronal In Vitro Model for Rett Syndrome Using a Direct Reprogramming Method. Anna Huber, Victoria Sarne, Alexander V Beribisky, Daniela Ackerbauer, Sophia Derdak, Silvia Madritsch, Julia Etzler, Sigismund Huck, Petra Scholze, Ilayda Gorgulu, John Christodoulou, Christian R Studenik, Winfried Neuhaus, Bronwen Connor, Franco Laccone, Hannes Steinkellner. Stem Cells Dev. 2024 Mar;33(5-6):128-142.doi: 10.1089/scd.2023.0233. Epub 2024 Feb 22.
-
TAT-MeCP2 protein variants rescue disease phenotypes in human and mouse models of Rett syndrome. Steinkellner H, Kempaiah P, Beribisky AV, Pferschy S, Etzler J, Huber A, Sarne V, Neuhaus W, Kuttke M, Bauer J, Arunachalam JP, Christodoulou J, Dressel R, Mildner A, Prinz M, Laccone F. Int J Biol Macromol. 2022 Apr 20;209(Pt A):972-983. doi: 10.1016/j.ijbiomac.2022.04.080. Online ahead of print. PMID: 35460749
- An electrochemiluminescence based assay for quantitative detection of endogenous and exogenously applied MeCP2 protein variants. Steinkellner H, Schönegger A, Etzler J, Kempaiah P, Huber A, Hahn K, Rose K, Duerr M, Christodoulou J, Beribisky AV, Neuhaus W, Laccone F. Sci Rep. 2019 May 28;9(1):7929. doi: 10.1038/s41598-019-44372-3.
- Detection of survival motor neuron protein in buccal cells through electrochemiluminescence-based assay. Steinkellner H, Etzler J, Gmeiner BM, Laccone F. Assay Drug Dev Technol. 2015 Apr;13(3):167-73. doi: 10.1089/adt.2015.635. Epub 2015 Apr 7.
- Identification and molecular characterisation of a homozygous missense mutation in the ADAMTS10 gene in a patient with Weill-Marchesani syndrome. Steinkellner H, Etzler J, Gogoll L, Neesen J, Stifter E, Brandau O, Laccone F. Eur J Hum Genet. 2015 Sep;23(9):1186-91. doi: 10.1038/ejhg.2014.264. Epub 2014 Dec 3.
- Mild overexpression of Mecp2 in mice causes a higher susceptibility toward seizures. Bodda C, Tantra M, Mollajew R, Arunachalam JP, Laccone FA, Can K, Rosenberger A, Mironov SL, Ehrenreich H, Mannan AU. Am J Pathol. 2013 Jul;183(1):195-210. doi: 10.1016/j.ajpath.2013.03.019. Epub 2013 May 15.